
RNA is crucial in regulating cellular processes, including gene expression, protein synthesis, and post-translational modifications. Understanding the functional dynamics of RNA is essential for advancing molecular biology and therapeutic research.
However, non-coding RNAs (ncRNAs), especially long non-coding RNAs (lncRNAs), have emerged as key players in cellular mechanisms despite ongoing debate about their functional significance. By studying RNA sequence, structure, and function, you gain deeper insights into cellular processes. These investigations are essential for uncovering the mechanisms of gene regulation, understanding disease pathways, and exploring the evolution of genetic systems.
High-throughput technologies like RNA sequencing (RNA-Seq) enable you to explore the human transcriptome in detail, providing researchers with a large amount of data. Bioinformatics also helps manage this complexity, offering computational methods to predict RNA structure, function, and interactions that are crucial for understanding their role in health and disease.
RNA Sequence Analysis Methods
Source: NIH
| While the protein-coding genes in the human genome have long been the focus of research, a deeper look reveals that the story is far more complex than initially thought. Around 1.2% of our genome codes for proteins, but a growing body of evidence suggests that a significant portion is transcribed into non-coding RNAs (ncRNAs), such as long non-coding RNAs (lncRNAs). |
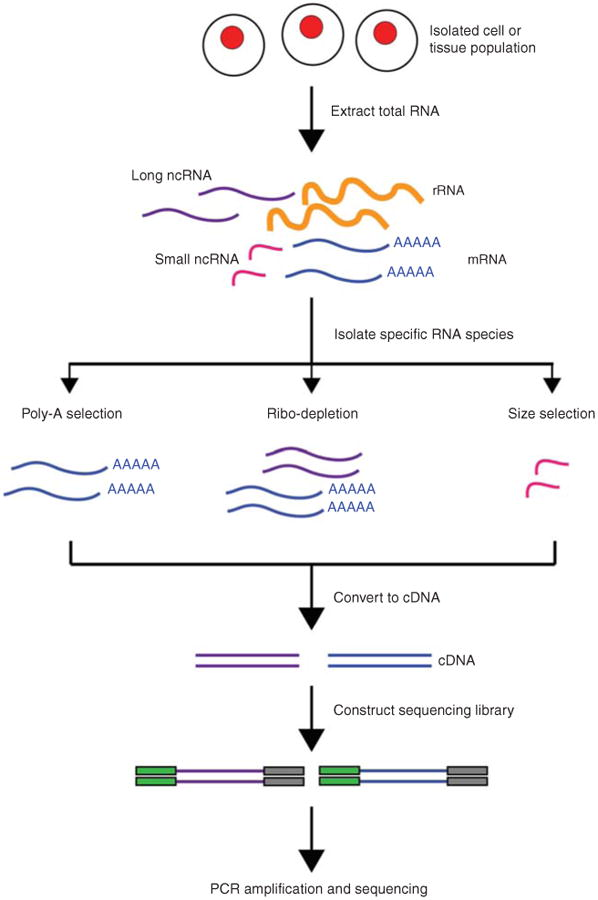
The RNA-Seq analysis pipeline starts with generating FASTQ files containing raw reads from next-generation sequencing (NGS). These reads are aligned to a reference genome, followed by the quantification of gene expression. Although RNA-Seq tools are now widely accessible, the method still presents unique computational challenges due to inherent biases in expression data.
1. Read Alignment
RNA-Seq read mapping is more complex than DNA sequencing because many reads span splice junctions. Standard tools like Bowtie and BWA cannot handle spliced transcripts. To address this, splicing-aware aligners such as GSNAP, MapSplice, STAR, and TopHat are used. These tools differ in performance, speed, and memory, so selecting the best one depends on the study’s objectives. Evaluations like RGASP3 help identify the most suitable tool based on alignment performance.
2. Transcript Assembly and Quantification
After aligning reads, they are assembled into transcripts. Most tools infer transcript models from read alignments, but de novo transcript reconstruction remains a challenge. Gene expression levels are quantified using tools like Cufflinks, FluxCapacitor, and MISO, which count reads aligned to full-length transcripts.
Alternatives like HTSeq count reads mapping to specific exons. To correct for biases like sequencing depth, read counts are normalized using metrics like RPKM or FPKM. Challenges arise when reads map to multiple isoforms. Some methods exclude non-unique reads, while others use likelihood models to determine the most likely isoform for each read.
3. miRNA Sequencing Analysis
miRNA sequencing follows similar methods to mRNA analysis. miRNAs are small and lack poly-A tails, making them more challenging to study. Tools like miRanalyzer and miRDeep help identify known miRNAs and predict new ones, as well as perform target prediction and expression analysis.
4. Quality Control and Technical Considerations
RNA-Seq analysis requires careful quality control at every step. Tools like FastQC and the FASTX toolkit assess sequence quality, checking for issues like base composition, adapter contamination, and sequencing errors. Trimming the ends of reads can correct mispriming errors.
It’s also important to assess mapping parameters like coverage bias and read distribution. Misalignments at splice junctions can occur, especially with incomplete reference annotations. Comparing DNA and RNA sequences can help identify sample issues like swaps or mixtures.
RNA-Seq is a powerful tool for gene expression analysis, but it comes with challenges like experimental design, alignment strategies, and quality control.
RNA Structure Analysis: Techniques and Biological Insights
Source: NIH
The analysis of RNA structure has evolved significantly with the introduction of sequence-independent chemistries and high-throughput software. These tools enable a deeper understanding of RNA’s role in various biological processes.
- Sequence-Independent Chemistries for RNA Structure Analysis
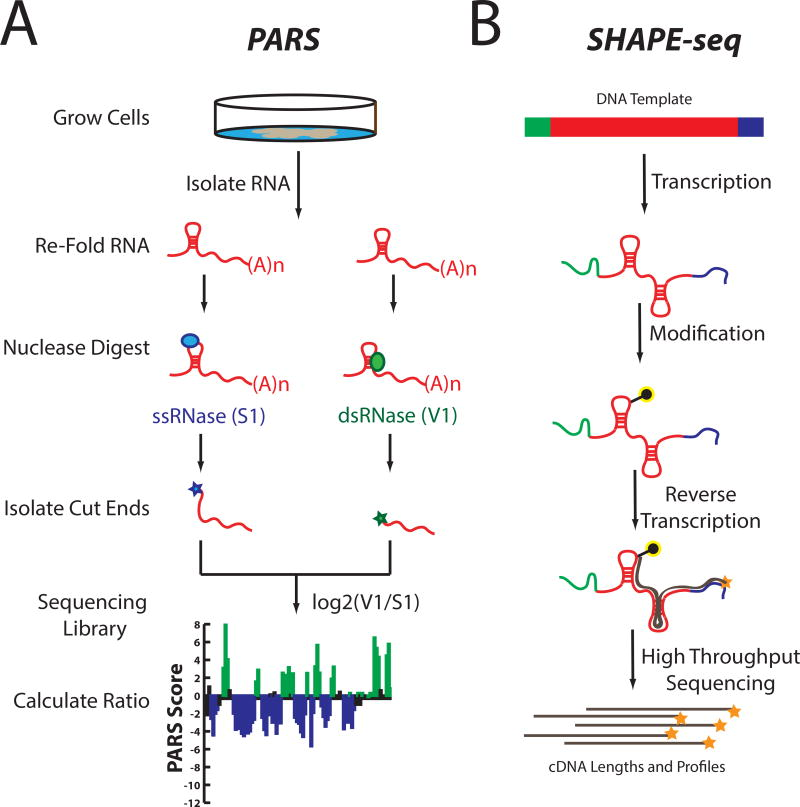
Traditional base-selective chemical reagents, such as DMS, have been fundamental in advancing our understanding of RNA structure-function relationships. However, to fully interrogate every nucleotide, multiple reagents are typically required. Techniques like in-line probing and SHAPE chemistry are particularly useful in this regard.
In-line probing measures local nucleotide flexibility and has proven valuable for developing secondary structure models for small RNAs. Additionally, RNases like E. coli RNase I and B. subtilis RNase J1, which act independently of sequence, provide another dimension to RNA analysis, making it possible to examine the local nucleotide environment at almost every position in RNA simultaneously.
- Software for High-Throughput RNA Structure Analysis
The development of high-throughput software solutions has dramatically streamlined the analysis of RNA structures. Traditionally, resolving and quantifying fragments from chemical probing experiments was time-consuming and labor-intensive. However, programs like the Semi-Automated Footprinting Analysis (SAFA) program have made this process much faster, reducing hours of work to just 20–60 minutes.
In addition, technologies like capillary electrophoresis have been incorporated into RNA structure analysis, enabling single-nucleotide resolution across large RNA sequences. Software systems such as Capillary Automated Footprinting Analysis (CAFA) and ShapeFinder are specifically designed to analyze RNA structure with high efficiency, offering distinct advantages depending on the RNA length and complexity.
- Secondary Structure Determination
A fundamental aspect of RNA structure analysis is understanding its secondary structure, which refers to the base pairing pattern. While comparative sequence analysis is the gold standard for this, it only applies to RNAs with homologs. Advances in dynamic programming algorithms for predicting RNA secondary structure have also made notable progress, though challenges remain, especially for larger RNAs.
One solution is integrating experimental chemical mapping data into these algorithms, which can improve prediction accuracy. For example, combining SHAPE reactivity data with free energy minimization algorithms has resulted in more accurate predictions for structures like the E. coli 16S ribosomal RNA, achieving up to 97% accuracy.
- Time-Resolved RNA Structure Analysis
Understanding RNA folding and the dynamics of RNA-protein complexes requires knowledge of their structural evolution over time. Many RNAs fold through complex pathways, often involving multiple stable intermediates and non-functional states. Time-resolved techniques, such as fast Fenton hydroxyl radical footprinting, have enabled researchers to capture RNA folding events in real time, spanning from milliseconds to minutes.
This approach has provided crucial insights into the folding mechanisms of various RNA structures, such as the RNase P RNA and riboswitch interactions, revealing the kinetics and conformational 5. The changes involved in RNA folding and assembly.
- Tertiary Structure Constraints
Many RNAs undergo complex tertiary structural interactions that are key to their biological functions in addition to secondary structures. Chemical mapping reagents can be used to infer these higher-order interactions by identifying nucleotides that are protected from cleavage, suggesting the presence of tertiary contacts.
Combining this with known high-resolution structures from NMR or X-ray crystallography allows for a more complete picture of RNA folding and function. Techniques like multiplexed hydroxyl radical cleavage analysis (MOHCA) and cross-linking studies have provided valuable through-space connectivity data, which is essential for refining three-dimensional models of RNA structures.
With these combined advancements in RNA structure analysis, researchers are now able to tackle increasingly complex biological questions, leading to deeper insights into RNA. Below, you will explore the functional analysis of RNA.
Functional Analysis of RNA in Poliovirus RNA Replication

Functional analysis of RNA refers to studying how RNA molecules work within the cell. It focuses on understanding the roles and activities of different types of RNA (such as messenger RNA, transfer RNA, and ribosomal RNA) in processes like protein synthesis, gene regulation, and RNA replication.
Below, you will explore in more depth the functional analysis of RNA in poliovirus RNA replication to understand the concept better:
- Poliovirus RNA Polymerase (3D)
Poliovirus uses a special protein called RNA polymerase (3D) to make copies of its RNA. This polymerase works by sticking to membranes in the cell, where it forms large groups (called oligomers) to help copy the virus’s RNA.
- Importance of Interface I and Interface II
The interface regions of the polymerase, particularly Interface I and Interface II, are essential for RNA binding and elongation. These interfaces are crucial for the polymerase to bind to RNA and make copies. When mutations damage these regions, the polymerase cannot bind to RNA as well or copy it efficiently.
- Polymerase Structure and RNA Binding
When it interacts with RNA, the polymerase can form large, ordered structures, like sheets or tubes. These structures help the polymerase work better, as they provide a surface for the RNA to bind to, making the replication process more efficient.
- Polymerase Action in the Infected Cell
In the infected cell, the polymerase sticks to special membranes and forms these large structures, helping the virus replicate. The polymerase’s ability to stick to the membrane and create these structures is essential for the virus to make more RNA and reproduce.
In simple terms, the polymerase works like a “machine” that forms big groups, sticks to cell membranes, and helps copy the virus’s RNA. If its structure is disrupted, it can’t work properly, which stops the virus from reproducing.
RNA-Protein Interactions in Gene Regulation and Cellular Function
Source: NIH
RNA and proteins are closely intertwined in cellular functions, often influencing each other’s lifecycle through physical interactions. mRNA carries the genetic instructions for protein synthesis, and non-coding regions of mRNA can regulate protein translation, localization, and interaction with other proteins. In return, proteins regulate RNA through processes like synthesis and degradation. These RNA-protein interactions (RPIs) are critical for maintaining cellular homeostasis, and disruptions can lead to diseases.
RNA-Centric vs Protein-Centric Methods
RNA-protein interaction methods can be divided into two types: RNA-centric and protein-centric.
- RNA-Centric Methods: These methods start with an RNA molecule and focus on identifying which proteins bind to it. Techniques like RNA immunoprecipitation (RIP), cross-linking and immunoprecipitation (CLIP), and RNA pulldown assays are standard RNA-centric methods.
- Protein-Centric Methods: These methods start with a protein of interest and aim to identify which RNAs it binds to. Methods include RNA-binding protein immunoprecipitation (RIP), RNA affinity purification (RAP), and proximity ligation assays (PLA).
Both approaches have unique advantages depending on the specific research question. Recent advancements in RNA-protein interaction studies have expanded the range of methods available. High-throughput sequencing technologies, along with bioinformatic tools, allow for better mapping of these interactions. Computational models can also predict RNA-protein binding sites, enhancing experimental work.
Now, as above, you have to uncover the RNA-protein interaction; below, you will dive into the bioinformatic challenges.
RNA Bioinformatic Challenges and Future Directions
Now, you have landed on the last section of this article, which is about the challenges and future directions of RNA bioinformatics and sequencing.
Source-NIH: Single‐cell RNA Sequencing Technology
1. Current limitations in RNA bioinformatics
Despite significant advances in RNA bioinformatics, several technical and computational challenges persist, particularly in the study of circular RNAs (circRNAs). These limitations hinder accurate detection, quantification, and interpretation of RNA species, especially in complex transcriptomes.
Addressing these issues is crucial for improving the reliability and reproducibility of RNA-based analyses in both basic and clinical research.
- Low Abundance of circRNAs: CircRNAs are often expressed at low levels in common cell lines (1-3% of mRNA levels), which makes their identification and quantification difficult.
- Identification Challenges in RNA-Seq: In single-end RNA-seq data, circRNAs are detected by reads aligned to the backsplice junction. However, sequencing biases can influence these junctional reads, and distinguishing between linear and circular isoforms is challenging.
- False-Positive Detection: CircRNA detection algorithms face issues with false positives, especially when sequencing errors or homology between exons lead to incorrect alignments. The algorithms used to detect circRNAs often apply thresholds (such as read counts) to minimize false positives, but this can lead to reduced sensitivity, especially for genes expressed at low levels.
2. Emerging technologies in RNA analysis
Emerging technologies in RNA analysis include single-cell RNA sequencing (scRNA-seq), which allows for the study of gene expression at the single-cell level, and long-read sequencing technologies like PacBio and Oxford Nanopore, which provide better insights into RNA isoforms and splicing patterns. Additionally, CRISPR-based RNA editing and RNA capture techniques are being developed to improve precision in RNA modification and detection. These advancements enhance the resolution, accuracy, and scope of RNA research.
Conclusion
This article helped you uncover RNA sequence structure and function, computational and bioinformatic methods. Along with this, the contribution and role of RNA in cellular processes, from its sequence and structure to its function and interactions with proteins. Through high-throughput RNA sequencing and bioinformatic methods, we can better understand RNA’s complex roles in gene regulation, disease mechanisms, and cellular processes.
As technology advances, so does the ability to study RNA at a deeper level, but challenges remain in bioinformatics, particularly with low-abundance RNA types like circRNAs. Continued research and development of more sophisticated computational tools are essential to overcome current limitations in RNA analysis. Integrating multi-omics data, improving algorithms for RNA isoform discrimination, and refining detection sensitivity will further enhance our ability to interpret RNA biology.
And with advanced future technologies and rapid development of modern tools and platforms like BioState AI, researchers can get comprehensive, high-quality RNA sequencing at an affordable price point. The combination of cost-efficiency and superior quality, with no worries about complexities and time-consuming aspects of experimental work, makes it a great option for you. Get Your Quote Now!
FAQs
Q: What are the methods to study RNA structure?
A: RNA structure can be studied using experimental techniques like SHAPE, DMS, and cryo-EM, along with computational tools such as RNAfold and RNAstructure. These methods help reveal secondary and tertiary conformations, aiding in functional annotation and interaction prediction.
Q: What are the different types of RNA structure and function?
A: RNA structures include primary (sequence), secondary (base-pairing, stems, loops), and tertiary (3D folding). Functions vary by type: mRNA carries coding information, tRNA and rRNA aid translation, while lncRNAs and circRNAs regulate gene expression and cellular signaling.
Q: What is the chemical structure and base composition of DNA and RNA?
A: Both DNA and RNA are nucleic acids composed of nucleotide monomers. DNA has deoxyribose sugar and uses thymine, while RNA has ribose and uses uracil. Both contain adenine, cytosine, and guanine, forming sequences that determine genetic and regulatory information.
Q: How to integrate different RNA-seq datasets?
A: Integration involves normalization (e.g., TPM, RPKM), batch effect correction (e.g., ComBat, Harmony), and metadata alignment. Tools like DESeq2, Seurat, and limma-voom support cross-dataset comparison for differential expression and clustering while accounting for platform and batch variability.
Q: What are the most optimized ways to predict plant lncRNA-mRNA interactions?
A: Combine expression correlation with computational tools like LncTar, RIblast, or IntaRNA. Use high-quality annotations and apply filters based on sequence complementarity, energy minimization, and co-expression across conditions. Integration with degradome or CLIP-seq data can improve confidence.