
If you’ve worked with RNA sequencing, you know that getting reliable data from challenging or degraded samples can be a real hurdle. Many standard RNA-Seq methods depend on high-quality RNA and often miss important transcripts when the sample isn’t ideal. This is a common pain point, especially when working with clinical samples, archived tissues, or rare cell populations.
Exome capture RNA-Seq is designed to address these issues. Instead of sequencing all RNA, this method targets only the coding regions, known as exons, where most disease-causing mutations and functional changes occur. As a result, it improves sensitivity for detecting gene expression changes, even in low-quality or fragmented RNA.
By not relying on poly-A tails, exome capture RNA-Seq works well with both high-quality and compromised RNA, a flexibility important for clinical and translational research involving variable sample integrity.
In this blog, we will discuss the fundamentals of exome capture RNA-Seq, including its working principles and how it differs from conventional RNA sequencing methods.
TL;DR
- Exome capture RNA-seq targets only coding regions (exons), making it ideal for detecting disease-relevant mutations and gene expression changes, especially in degraded or clinical samples.
- Unlike standard RNA-seq, it does not require poly-A tails, so it works with fragmented or low-quality RNA, such as FFPE tissues and rare cell populations.
- By focusing sequencing on exons, it reduces both sequencing costs and the burden of data analysis, while increasing sensitivity for important transcripts.
- Technical hurdles exist, like off-target capture and uneven coverage, but new probe designs and optimized workflows are improving reliability.
What is Exome Capture
Exome capture, a crucial step in Whole Exome Sequencing (WES), focuses on selectively enriching the protein-coding regions (exons) of the transcriptome. These regions contain the majority of known disease-related mutations and are central to understanding gene expression and function.
Key Advantages of exome capture

- Cost-Effectiveness: Focuses sequencing on the protein-coding regions, reducing sequencing depth and cost compared to whole transcriptome sequencing (total RNA-Seq).
- High Sensitivity for Coding Regions: Provides higher coverage for exonic regions, improving the detection of low-abundance transcripts and variants within these critical areas.
- Suitable for Degraded Samples: Does not rely on poly(A) tails, making it ideal for challenging samples like FFPE tissues, where RNA is often fragmented.
- Improved Discovery Power: By concentrating reads on high-value content, it enhances the ability to discover novel transcripts, alternative splice variants, and gene fusions within the exome.
- Reduced Data Storage and Analysis Burden: Generates less data than total RNA-Seq, simplifying data storage and bioinformatics analysis.
Exome capture RNA-Seq is widely used in cancer genomics, disease research, and studies requiring high sensitivity from challenging samples, making it a practical tool for use when you are working with diverse RNA sources.
To help you make informed decisions for your research, let’s examine how exome capture RNA-Seq stacks up against conventional RNA-Seq.
Exome Capture RNA-Seq vs Conventional RNA-Seq: Key Differences
Traditional RNA-Seq captures the entire transcriptome by sequencing all RNA molecules present in a sample, providing a comprehensive view of gene expression across coding and non-coding regions.
Understanding the differences between conventional RNA-Seq and exome capture RNA-Seq becomes critical when you need to optimize your research budget while maintaining the depth of analysis your study demands.
| Parameter | Conventional RNA-Seq | Exome Capture RNA-Seq |
| Target Coverage | Sequences the entire transcriptome, including introns, UTRs, and intergenic regions | Focuses specifically on protein-coding exons and splice junctions |
| Library Preparation | Uses random hexamer or oligo-dT priming for cDNA synthesis | Employs targeted enrichment probes after initial cDNA synthesis |
| Sequencing Depth | Requires 20-50 million reads for standard differential expression analysis | Achieves comparable results with 10-20 million reads due to focused targeting |
| Cost per Sample | Higher due to broader coverage requirements | Lower cost per sample with concentrated sequencing effort |
| Variant Detection | Identifies variants across the entire transcript length, including splice sites | Optimized for coding variant detection with enhanced sensitivity |
| Novel Transcript Discovery | Excellent for identifying new isoforms and non-coding transcripts | Limited to known exonic regions, misses novel transcripts |
| Splicing Analysis | Provides comprehensive splice junction analysis | Focuses on known splice sites with targeted probe design |
| Data Complexity | Generates larger datasets requiring more computational resources | Produces focused datasets with reduced computational burden |
| Clinical Applications | Suitable for basic research and discovery-phase studies | Preferred for clinical diagnostics and targeted therapeutic research |
| Coverage Uniformity | Variable coverage across transcripts due to random sampling | Enhanced uniformity across targeted exonic regions |
Exome capture RNA-Seq delivers targeted efficiency when your research focuses on protein-coding variants and clinical applications, while conventional RNA-Seq remains essential for comprehensive transcriptome discovery.
Your choice between these methods should align with your specific research objectives, budget constraints, and the depth of novel transcript discovery your study requires.
Understanding these differences sets the stage for a closer look at how exome capture is performed in the lab, from probe design to the technology that makes targeted sequencing possible.
Explanation of Exome Capture RNA-seq Methods
Exome capture, primarily employed in DNA sequencing, specifically targets and analyzes the exome, which comprises the protein-coding regions of the genome. This constitutes approximately 2% of mammalian DNA. This targeted approach typically utilizes liquid-phase hybrid capture technology.
Here are the fundamental principles of hybridization capture:
Base Pairing and Probe Design
Hybridization capture is based on the principle of base pairing, where complementary nucleic acid strands bind together through hydrogen bonds. To capture specific RNA sequences, scientists design probes (also known as baits or oligonucleotides) that are complementary to the exonic RNA of interest.
In RNA-Seq, the choice of probe chemistry has evolved over time. Early methods often used biotin-labeled RNA probes for hybridization. However, modern approaches have shifted to using DNA probes, and here’s why:
- RNA is less stable than DNA, making it prone to degradation during the capture process.
- RNA-Seq workflows involve reverse-transcribing RNA into cDNA, which is more stable than RNA. Therefore, using DNA probes that are complementary to the cDNA library offers better stability and robustness during hybridization.
- DNA probes are also less likely to degrade compared to RNA, ensuring more consistent performance across different experiments.
This shift to DNA probes has significantly improved the capture process, making it more reliable and efficient, especially when working with RNA-derived samples.
Key Characteristics of Effective Probes
For probes to work efficiently, they must meet certain criteria:
- Single-Stranded: The probe must be single-stranded for proper hybridization.
- High Specificity: The probe should bind only to its intended target, avoiding any cross-hybridization with non-target sequences.
- No Self-Annealing: The probe should not bind to itself, as self-annealing would reduce the amount of probe available for hybridization with the target sequence.
To ensure high specificity, probes typically need to be at least 18 bases long. This length reduces the likelihood of non-specific binding in complex genomes. Specialized software such as Probefinder, MiPRIME, etc., assists researchers in selecting probe sequences that minimize secondary structures and evaluate the likelihood of non-specific hybridization with other genomic regions.
The Role of Biotinylated Probes and Magnetic Beads
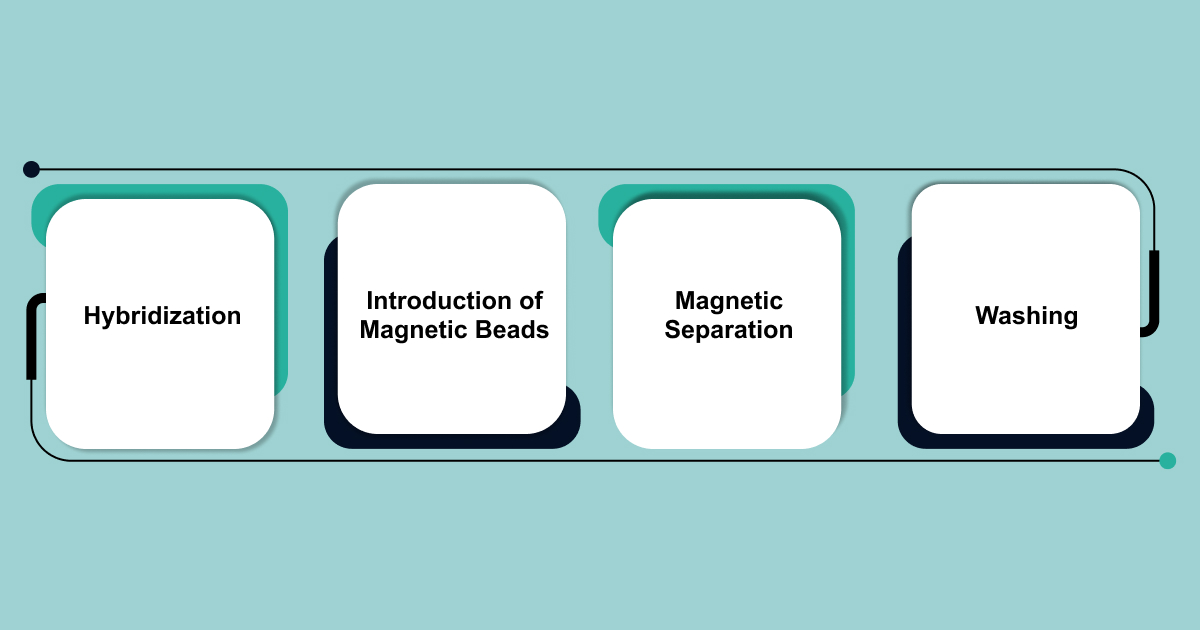
In the liquid-phase hybrid capture process, researchers attach biotin molecules to capture probes. Biotin serves as a tag that makes isolation easier. Here’s how the process works:
- Hybridization: Biotinylated probes bind to complementary target RNA or cDNA sequences.
- Introduction of Magnetic Beads: Streptavidin-coated magnetic beads are added to the solution. Streptavidin has a strong affinity for biotin, so it binds tightly to the biotinylated probes.
- Magnetic Separation: A magnetic field is applied, pulling the magnetic beads—and the attached target RNA/cDNA fragments—toward one side of the vessel. This step helps separate the target sequences from non-target material.
- Washing: Researchers perform a series of washing steps to remove unbound, non-target material.
This method allows for the efficient capture and isolation of specific target sequences, improving the accuracy of RNA or cDNA enrichment.
With the methodology in mind, let’s walk through the full workflow of exome capture RNA-Seq, step by step, from sample preparation to sequencing, to see how each part contributes to high-quality results.
Comprehensive Workflow of Exome Capture RNA-Seq
Exome capture RNA-Seq involves a series of carefully coordinated steps, each designed to ensure the extraction of high-quality, targeted transcriptomic data. From initial sample handling to sequencing, these steps must be executed precisely for optimal results.
- Sample Preparation
The process begins with RNA extraction from various biological sources, such as:
- Whole blood
- Peripheral blood mononuclear cells (PBMCs)
- Freshly frozen tissues
- Formalin-fixed paraffin-embedded (FFPE) samples
- Plasma
- Amniotic fluid cells
RNA is inherently unstable, so it is crucial to preserve its integrity. For FFPE samples, which often suffer from RNA fragmentation and chemical modifications due to the fixation process, specialized RNA isolation kits (e.g., Qiagen RNeasy FFPE kit) are necessary. Quality is assessed using metrics like:
- RNA Integrity Number (RIN)
- DV200 (percentage of RNA fragments longer than 200 nucleotides)
Some kits support samples with a RIN >2 or DV200 >30, indicating the sample’s suitability.
Next, the RNA undergoes fragmentation into smaller pieces. Researchers then reverse transcribe these RNA fragments into complementary DNA (cDNA). This conversion is essential, as cDNA is more stable and better suited for further processing.
Stranded RNA-Seq protocols incorporate modified dUTP nucleotides during second-strand cDNA synthesis to preserve strand information, crucial for accurate gene expression and isoform analysis.
- Library Construction
Once cDNA is synthesized, researchers repair the ends of the cDNA fragments, creating blunt ends that are suitable for adapter ligation.
- They then ligate specific adapter sequences to both ends of the cDNA fragments.
- These adapters are essential for PCR amplification and binding to the sequencing flow cell.
- After ligation, an initial round of PCR amplification increases the cDNA library’s quantity, ensuring enough material for capture and sequencing.
- Some advanced kits, like IDT’s xGen RNA Library Prep kits, integrate Adaptase™ technology, which optimizes the process, particularly for low-quality or low-input samples.
- Targeted Exome Capture and Enrichment
The key to successful exome capture lies in the design of the probes.
- Researchers design biotinylated oligonucleotide probes (baits) to target specific exonic regions within the cDNA library.
- The probe design software ensures high specificity and minimizes off-target binding, accounting for any secondary structures or problematic sequences.
- For degraded RNA samples, a higher probe concentration at multiple positions along the target transcript improves capture efficiency, even for fragmented molecules.
- The probes can be designed for tiling, covering large regions, or overlapping, offering extra coverage at fragment ends to avoid gaps in sequencing.
- This is followed by a hybridization reaction, where the cDNA library is mixed with the biotinylated probes.
Purification and Elution
After hybridization, researchers introduce streptavidin-coated magnetic beads into the mixture.
- The biotinylated probes bind to the target cDNA fragments, and these complexes attach to the magnetic beads.
- Applying a magnetic field separates the bead-bound target complexes from the unbound cDNA fragments.
- Multiple stringent washing steps remove any non-specific bindings, ensuring high purity of the enriched exome.
- Finally, the captured cDNA fragments are eluted from the magnetic beads.
- Distilled water is often sufficient for this, but specialized commercial kits may require specific solutions for better recovery.
The reaction occurs overnight under controlled temperature and salt conditions, promoting specific binding between the probes and the cDNA fragments.
- Post-Capture PCR Amplification and Sequencing
Following elution, the enriched cDNA library is further amplified via PCR to generate sufficient material for high-throughput sequencing. The number of PCR cycles is carefully optimized to avoid over-amplification, which can introduce biases.
- Library Quality Control: The final library is rigorously assessed for its quality, quantity, and size distribution using techniques like:
- Qubit or NanoDrop: For concentration measurement.
- Bioanalyzer or Fragment Analyzer: To check the size distribution and confirm the presence of adapter-ligated fragments within the expected range.
Researchers then load the amplified and enriched libraries onto a Next-Generation Sequencing (NGS) platform.
- High-Throughput Sequencing (NGS):
- NGS platforms such as Illumina’s HiSeq, NextSeq, or NovaSeq systems are used.
- NGS involves coupling the exome sample to a solid support, like a flow cell, and performing in situ PCR duplication to amplify the signal from each cluster.
- Illumina’s sequencing by synthesis (SBS) chemistry is the most widely adopted NGS technology globally.
- This process generates strings of continuous sequence data in the form of “reads,” which are commonly short reads (typically ≤ 300 base pairs)
- Massively parallel sequencing generates millions of short reads (e.g., 50-150 bp paired-end reads) corresponding to the captured exonic regions.
Even with a well-designed workflow, exome capture RNA-Seq comes with its own set of technical challenges. Recognizing these issues is key to getting reliable data and making informed decisions.
Technical Challenges in Exome Capture RNA Sequencing
Exome capture RNA-seq offers a powerful combination of targeted exome capture and RNA-seq’s quantitative capabilities. However, several technical challenges complicate its implementation.
Here are the key challenges you can normally face in exome capture:
- Gene Family Distinction: Probes must distinguish closely related gene family members while maintaining strong binding affinity across diverse expression levels.
- Off-Target Capture: Pseudogenes and repetitive elements can bind to probes, leading to inaccurate results and skewed quantification.
- Uneven Coverage: Degraded RNA results in uneven coverage, which complicates downstream analysis.
- 5′ Bias: The typical 5′ bias in RNA-seq becomes more pronounced after capture, especially in longer transcripts.
- FFPE Samples: Formalin-fixed samples present unique issues. Crosslinking fragments RNA in unpredictable ways, making uniform capture difficult.
- Capture Efficiency Variability: Capture efficiency varies due to secondary structure, GC content, and transcript abundance. Highly expressed genes may saturate probes, while lowly expressed genes may not be detected.
- Biases in Normalization: Traditional RNA-seq normalization often fails because the captured transcript subset does not represent the full transcriptome. Researchers must develop capture-specific strategies to correct for these biases.
- Timing Challenges: Extended hybridization improves capture efficiency but can also lead to more off-target binding, requiring careful optimization.
- Differential Expression Analysis: Comparing captured samples to whole transcriptome RNA-seq data complicates differential expression analysis.
- Additional Costs: The cost of reagents and labor can offset the savings from reduced sequencing costs.
- Multiplexing: While multiplexing helps lower per-sample costs, it introduces variability in capture efficiency across samples.
While exome capture RNA-seq offers many advantages, overcoming these technical challenges is essential for optimizing results and ensuring the accuracy and efficiency of the workflow. Here’s where Biostate AI shines.
How Biostate AI Can Become a Solution To Overcome These Challenges
The technical challenges in exome capture can slow progress, increase costs, and add complexity to the research process. You need a streamlined solution that ensures high-quality results without the overwhelming technical demands.
Biostate AI offers a comprehensive solution to these challenges, simplifying the RNA sequencing process from start to finish. With its full-service approach, we take care of every step, from sample collection to the final insights, so researchers can focus on their core research.
Key features of Biostate AI’s RNA sequencing solution:
- Unbeatable Pricing: Start getting high-quality sequencing results for as low as $80 per sample.
- Rapid Turnaround: Receive your results in just 1-3 weeks.
- Complete Transcriptome Insights: Comprehensive RNA-Seq covering both mRNA and non-coding RNA.
- AI-Driven Analysis: Access powerful, intuitive insights with OmicsWeb AI.
- Minimal Sample Requirement: Process samples as small as 10µL of blood, 10ng of RNA, or 1 FFPE slide.
- Low RIN Compatibility: Compatible with RNA samples having a RIN as low as 2 (vs. the typical ≥5).
By combining affordability with high-quality sequencing and AI-driven analysis, Biostate AI makes RNA sequencing easier, more accessible, and more insightful than ever before.
Final Words
Exome capture RNA-seq is a powerful tool for researchers who need to study gene expression in challenging or degraded samples. This method saves time and money while giving you the sensitivity needed for clinical and disease research.
However, exome capture RNA-seq is not without its difficulties. Issues like off-target binding, uneven coverage, and complex data analysis can slow down your research and increase costs. Managing these technical challenges often requires special skills and extra resources.
That’s why having a reliable partner matters. Biostate AI offers a complete RNA sequencing solution starting at $80 per sample. With us, you don’t have to worry about complicated protocols or expensive lab work. You get reliable, accurate results at a price that fits your budget.
Get in touch with us today and see how our complete solution can help you achieve your research goals without the hassle.
FAQ
- How much starting material is required for exome capture RNA sequencing?
Exome capture RNA-seq can work with limited starting material. Some protocols accommodate as little as 5–25 ng of total RNA for low-input applications, though standard recommendations are often higher (e.g., 500 ng).
- Will low RIN or fragmented RNA affect my results in exome capture?
Exome capture RNA-seq is well-suited for degraded or fragmented RNA. Unlike poly(A) selection, exome capture uses probes that can bind throughout the transcript, allowing for reliable results even from low RIN or fragmented samples (such as those from FFPE tissue). However, extremely degraded RNA can still impact transcript coverage and quantification, so some caution is warranted.
- How does RNA quality (RIN) affect exome capture RNA sequencing results?
For standard RNA-seq, a higher RIN (≥7–8) is preferred for optimal results. Lower RIN can reduce the number of uniquely mapped reads and affect gene coverage, potentially leading to false positives in differential expression analysis. Exome capture protocols are more tolerant of low RIN compared to poly(A) selection, providing accurate gene expression data even from degraded RNA, but very low RIN may still introduce some bias.
- What is the typical sequencing depth needed for reliable results?
Typical sequencing depth for exome capture RNA-seq is 30–50 million reads per sample for robust gene expression analysis. For the detection of gene fusions or low-abundance transcripts, higher depth may be required.