
Unlocking the insights hidden within RNA sequencing data is a race against time and complexity. As next-generation sequencing technologies generate ever-larger datasets, researchers must navigate a maze of quality checks, read alignment, and data interpretation to reach meaningful results quickly. Fast RNA sequencing analysis means making smart choices at every step, from optimizing your computational setup to selecting the right bioinformatics tools and workflows.
We know that managing massive datasets and the intricate steps of RNA-seq analysis can often feel overwhelming, especially when tight deadlines and high research stakes are involved. You’re not alone in these challenges, and with the right strategies, faster, more reliable results are within reach.
In this blog, we dive into fast RNA sequencing analysis tips and proven strategies that empower you to accelerate your RNA-seq analysis, minimize bottlenecks, and extract robust biological insights with confidence. These actionable recommendations will make your analysis faster, smoother, and more effective.
TL;DR
Fast RNA-seq analysis requires strategic planning and the right tools:
- Plan ahead: Choose paired-end sequencing for complex studies, target 20-30M reads for standard expression profiling, use 3+ biological replicates
- Prioritize RNA quality: Aim for RIN >8, but modern platforms like Biostate AI can work with RIN as low as 2
- Optimize library prep: Use mRNA-seq for focused studies, total RNA-seq for comprehensive analysis including non-coding RNAs
- Implement rigorous QC: Use FastQC/MultiQC immediately, remove contaminants (rRNA, mitochondrial sequences) to improve sensitivity
- Leverage automated pipelines: Use STAR aligner + Salmon quantification, implement cloud-based solutions for scalability
- Choose integrated platforms: Biostate AI offers end-to-end solutions ($80/sample, 1-3 week turnaround) with AI-powered analysis through OmicsWeb
- Document everything: Track software versions and parameters for reproducibility
Key metrics for success: 95% of genes detected with 20M reads, 89% accuracy in disease prediction with proper AI models, 3-5x improvement in gene detection with proper sample prep.
What is RNA Sequencing?
RNA sequencing (RNA-seq) is a powerful genomic technique that captures and quantifies the complete set of RNA transcripts produced by a genome at any given moment.
- RNA molecules are first converted into complementary DNA (cDNA) libraries.
- These cDNA libraries are sequenced using next-generation sequencing (NGS) platforms, generating millions of short DNA reads.
- The resulting reads are computationally assembled and aligned to a reference genome, allowing for detailed analysis of gene expression and transcript structure.
Key Advantages of RNA-seq
- Unbiased Detection: Captures both known and novel transcripts, including rare and low-abundance RNAs.
- High Sensitivity & Precision: Quantifies gene expression levels with remarkable accuracy.
- Splice Variant Identification: Detects alternative splicing events and isoforms.
- Dynamic Range: Offers a broader dynamic range compared to microarrays, enabling detection of both highly and weakly expressed genes.
Computational Challenges in Modern RNA-seq Workflows
The RNA sequencing market has experienced explosive growth, with the global NGS-based RNA-sequencing market valued at USD 3.92 billion in 2024 and projected to grow at a CAGR of 17.3% from 2025 to 2030.
However, modern RNA-seq workflows face several computational challenges that directly impact analysis speed. These are:
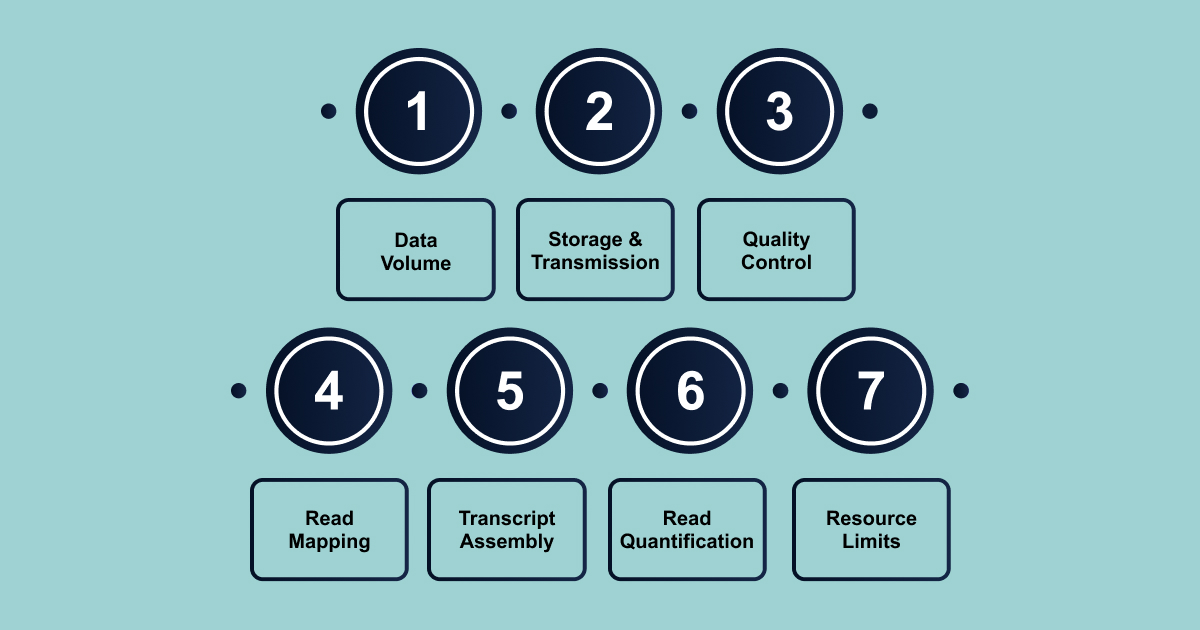
- Large Data Volume: Processing billions of sequencing reads requires significant computational power and time.
- Data Storage and Transmission: Handling and moving massive datasets can create bottlenecks, slowing down the entire workflow.
- Quality Control: Running comprehensive quality checks on large datasets can be time-consuming.
- Read Mapping: Aligning millions or billions of short reads to a reference genome is computationally intensive and often the slowest step.
- Transcript Assembly: Reconstructing transcripts from short reads, especially in complex transcriptomes, adds to processing time.
- Read Quantification: Accurately counting and assigning reads to genes or transcripts requires efficient algorithms to avoid delays.
- Resource Limitations: Limited memory and CPU resources can significantly slow down each stage of the analysis.
Addressing these speed-related challenges is essential for researchers who want to extract insights efficiently and make the most of their sequencing investments. That’s why the next section focuses on essential tips and practical strategies specifically designed to accelerate RNA sequencing analysis.
Fast RNA Sequencing Analysis Tips for Accelerating RNA Sequencing Analysis
The computational challenges in RNA-seq analysis directly impact the speed of the workflow. Given the significant computational hurdles that can slow down RNA-seq analysis, it’s clear that simply following standard workflows may not be enough to keep pace with modern research demands. Here are some fast RNA sequencing analysis tips that can help you improve the speed of your workflow.
1. Carefully Plan Your Experimental Design
Effective experimental design forms the foundation of efficient RNA sequencing analysis. Researchers who invest time in thorough planning significantly reduce downstream computational bottlenecks and avoid costly re-sequencing experiments.
a. Choosing Between Paired-End and Single-End Sequencing
Single-end sequencing reads DNA fragments from one direction, making it faster and more cost-effective for basic gene expression studies. This approach works well when you need to quantify known transcripts in well-annotated genomes. However, paired-end sequencing reads fragments from both directions, providing superior accuracy for detecting splice junctions, novel transcripts, and structural variants.
Companies like Illumina have demonstrated that paired-end sequencing improves mapping accuracy by 15-20% compared to single-end approaches, particularly for detecting alternative splicing events. For most transcriptomic studies, the additional cost of paired-end sequencing justifies the improved data quality and reduced analysis time.
b. Selecting Appropriate Sample Types and Conditions
Your choice of sample type directly impacts analysis complexity and processing time. Fresh-frozen tissues provide the highest RNA quality but require immediate processing. Formalin-fixed paraffin-embedded (FFPE) samples offer convenience for retrospective studies but introduce RNA fragmentation that complicates analysis.
For example, blood samples present unique challenges due to high globin mRNA content, which can comprise up to 80% of total mRNA. Implementing globin depletion during library preparation reduces sequencing waste and improves gene detection sensitivity.
c. Determining Optimal Sequencing Depth and Biological Replicates
Insufficient sequencing depth leads to poor detection of low-abundance transcripts, while excessive depth wastes resources without proportional benefits. For standard mRNA profiling, target 20-30 million mapped reads per sample.
Specialized applications require different depths. For example, small RNA sequencing needs only 5-10 million reads, while comprehensive isoform analysis may require 100+ million reads.
Biological replicates significantly impact statistical power and analysis reliability. The Genotype-Tissue Expression (GTEx) project demonstrated that three biological replicates detect 80% of differentially expressed genes, while six replicates achieve 95% detection efficiency.
2. Ensure High-Quality RNA Extraction
RNA quality directly determines the success of your entire sequencing experiment. High-quality RNA extraction accelerates downstream analysis by reducing computational complexity and improving data reliability.
a. Understanding RNA Integrity Numbers (RIN)
The RNA Integrity Number (RIN) provides a standardized measure of RNA degradation, ranging from 1 (completely degraded) to 10 (intact). Traditional protocols recommend RIN values above 8 for optimal results, but recent advances in library preparation have extended compatibility to lower-quality samples.
Biostate AI‘s innovative approach demonstrates how modern RNA sequencing can accommodate samples with RIN values as low as 2, compared to the typical requirement of RIN ≥ 5.
This breakthrough enables researchers to analyze valuable clinical samples that would otherwise be unusable, expanding research possibilities while maintaining data quality.
b. Minimizing Batch Effects Through Simultaneous Processing
Batch effects introduce unwanted variation that complicates statistical analysis and reduces reproducibility. Process all samples from a single experiment simultaneously using identical reagent lots, equipment, and personnel.
When simultaneous processing proves impossible, randomize sample processing order across experimental conditions. Implementing proper sample handling protocols reduces this variation and simplifies downstream analysis.
c. Preventing RNA Degradation During Sample Collection
RNA degradation begins immediately upon sample collection due to ubiquitous RNase activity. Implement rapid RNase inactivation strategies appropriate for your sample type. For tissue samples, snap-freezing in liquid nitrogen provides immediate preservation. Blood samples benefit from specialized collection tubes containing RNA stabilization reagents.
3. Prevent RNA Degradation Immediately
Time-sensitive RNA preservation directly impacts analysis speed and accuracy. Implementing proper preservation protocols prevents degradation-related artifacts that complicate computational analysis.
a. Rapid RNase Inactivation Strategies
RNases remain active even at low temperatures, making immediate inactivation crucial. Use RNase inhibitors during sample processing and maintain samples at -80°C for long-term storage. Avoid repeated freeze-thaw cycles, which fragment RNA and introduce analysis artifacts.
b. Specialized Sample Processing Techniques
Blood samples require DNase treatment to remove genomic DNA contamination that interferes with RNA quantification. Consider implementing globin mRNA depletion or ribosomal RNA removal to enhance gene detection sensitivity and maximize sequencing efficiency.
These preprocessing steps reduce computational burden by eliminating non-informative sequences and improving the signal-to-noise ratio in your data. Companies like Thermo Fisher Scientific report 3-5 fold improvements in gene detection sensitivity following proper sample preparation.
4. Optimize Library Preparation Strategy
Library preparation strategy significantly influences analysis complexity and processing time. Choosing appropriate methods based on your research objectives streamlines downstream computational workflows.
a. Selecting mRNA vs. Total RNA Library Preparation
mRNA library preparation uses poly(A) selection to enrich protein-coding transcripts, reducing library complexity and accelerating analysis. This approach works well for gene expression studies focused on coding sequences but misses important regulatory non-coding RNAs.
Total RNA library preparation captures all RNA species, including microRNAs, long non-coding RNAs, and ribosomal RNAs. While this approach provides comprehensive transcriptome coverage, it increases computational requirements and analysis time.
b. Adapting Methods for Sample Quality
Degraded samples require specialized library preparation protocols that accommodate fragmented RNA. Recent innovations in library preparation have enabled the successful sequencing of heavily degraded samples, including ancient DNA specimens and archived clinical samples.
Biostate AI‘s technology exemplifies this advancement by processing samples as small as 10µL blood, 10ng RNA, or single FFPE slides while maintaining analytical accuracy. This capability accelerates research timelines by eliminating sample re-collection and re-processing steps.
5. Select Appropriate Sequencing Depth and Read Length
Optimal sequencing parameters balance data quality with analysis speed and cost-effectiveness. Proper parameter selection reduces unnecessary computational overhead while ensuring sufficient coverage for your research objectives.
a. Matching Parameters to Experimental Goals
Standard gene expression profiling requires 25-60 million reads per sample for comprehensive transcriptome coverage. Small RNA sequencing needs only 5-10 million reads due to the limited diversity of small RNA species. Single-cell RNA sequencing optimizes for cell number rather than read depth, typically using 50,000-100,000 reads per cell.
Read length selection depends on your analytical goals. Single-end 50bp reads suffice for basic expression quantification, while paired-end 150bp reads improve isoform detection and novel transcript discovery.
b. Balancing Coverage and Cost
The Broad Institute’s analysis of over 10,000 RNA-seq samples revealed that 20 million reads capture 95% of expressed genes in human tissues. Additional sequencing depth provides diminishing returns for most applications, making 20-30 million reads optimal for cost-effective analysis.
However, specialized applications may require deeper sequencing. Allele-specific expression analysis needs 50-100 million reads to detect subtle differences, while comprehensive isoform analysis may require 200+ million reads per sample.
6. Perform Rigorous Quality Control on Raw Reads
Immediate quality control assessment accelerates downstream analysis by identifying and resolving data quality issues early in the workflow. Implementing systematic QC protocols prevents time-consuming troubleshooting later.
a. Essential QC Metrics and Tools
FastQC provides a rapid assessment of raw read quality, identifying common issues like adapter contamination, sequence duplication, and quality score degradation. MultiQC aggregates results across multiple samples, enabling batch-level quality assessment.
Key metrics include per-base sequence quality (should remain above Q20), sequence length distribution (should match expected read length), and adapter content (should be minimal). Address quality issues through appropriate preprocessing steps before alignment.
b. Automated QC Workflows
Automated QC pipelines reduce manual intervention and accelerate analysis. Tools like Trim Galore! automatically detect and remove adapter sequences while performing quality trimming based on user-defined parameters.
Leading sequencing centers report 40-60% reduction in analysis time through automated QC implementation. These workflows also improve reproducibility by standardizing quality assessment across projects and personnel.
7. Remove Contaminants and Unwanted RNA Species
Contamination removal improves analysis efficiency by reducing computational burden and enhancing signal detection for genes of interest. Strategic filtering approaches maximize sequencing value while minimizing processing time.
a. Identifying and Removing Common Contaminants
Ribosomal RNA typically comprises 80-90% of total cellular RNA but provides limited information for most studies. Ribosomal RNA depletion during library preparation or computational removal during analysis significantly improves gene detection sensitivity.
Mitochondrial RNA represents another abundant contaminant that can dominate sequencing reads. Computational filtering removes mitochondrial sequences while preserving nuclear gene expression data for analysis.
b. Optimizing Sequence Utilization
Effective contamination removal increases the proportion of informative reads from 10-20% to 60-80% in typical experiments. This improvement reduces required sequencing depth and accelerates downstream analysis while maintaining statistical power.
For example, proper contamination removal enables detection of additional genes per sample compared to unfiltered data, significantly enhancing analytical value.
8. Use Robust Computational Pipelines
Standardized computational pipelines ensure reproducible, efficient analysis while reducing manual intervention requirements. Well-designed workflows automate repetitive tasks and implement best practices throughout the analysis process.
a. Selecting Appropriate Alignment and Quantification Tools
STAR aligner provides fast, accurate alignment for mammalian genomes, typically processing samples 5-10 times faster than older alignment tools. Salmon offers ultra-fast transcript quantification through k-mer-based pseudo-alignment, reducing processing time from hours to minutes.
Modern pipelines integrate these tools seamlessly, automatically selecting optimal parameters based on sample characteristics and experimental design. This automation reduces analysis time while maintaining accuracy.
b. Cloud-Based Analysis Solutions
Cloud computing platforms enable scalable analysis of large RNA-seq datasets without local infrastructure investment. Amazon Web Services, Google Cloud Platform, and Microsoft Azure offer pre-configured bioinformatics environments that accelerate analysis deployment.
For example, Biostate AI‘s OmicsWeb platform exemplifies next-generation analysis solutions by providing comprehensive, intuitive data analysis without requiring coding expertise. The platform combines robust data storage with automated analysis workflows, making complex datasets accessible to researchers regardless of computational background.
c. AI-Enhanced Analysis Capabilities
Artificial intelligence transforms RNA sequencing analysis by automating complex interpretation tasks and providing intuitive data exploration interfaces. AI copilot systems enable researchers to analyze data using natural language queries, dramatically reducing the barrier to sophisticated analysis.
9. Interpret Results with Context and Biological Insight
Meaningful interpretation requires integration of statistical results with biological knowledge and experimental context. This integration transforms raw data into actionable scientific insights while maintaining analytical rigor.
a. Functional Annotation and Pathway Analysis
Gene Ontology (GO) enrichment analysis identifies biological processes and molecular functions represented in your differentially expressed gene sets. KEGG pathway analysis maps expression changes onto known biochemical pathways, revealing system-level effects of experimental perturbations.
Modern annotation tools provide real-time access to continuously updated biological databases, ensuring interpretation reflects current scientific knowledge. Automated annotation pipelines integrate multiple databases for comprehensive functional characterization.
b. Integrating Multi-Omics Data
RNA sequencing data gains additional context when integrated with other omics datasets. Combining RNA-seq with DNA methylation, chromatin accessibility, or proteomics data provides mechanistic insights into gene regulation and cellular function.
The ENCODE project demonstrated that multi-omics integration reveals regulatory relationships invisible in single-dataset analyzes. This comprehensive approach accelerates hypothesis generation and experimental design for follow-up studies.
c. Clinical Translation and Disease Applications
RNA sequencing increasingly supports clinical decision-making through disease classification, prognosis prediction, and treatment selection. Machine learning approaches trained on large clinical datasets can predict treatment outcomes with remarkable accuracy.
Biostate AI‘s Disease Prognosis AI exemplifies this application, achieving 89% accuracy in predicting drug toxicity and 70% accuracy in therapy selection for acute myeloid leukemia. These capabilities transform existing clinical samples into predictive disease models, accelerating personalized medicine development.
10. Document and Share Your Workflow
Comprehensive documentation ensures reproducibility and facilitates collaboration while accelerating future analyzes. Systematic record-keeping prevents time-consuming reconstruction of successful analysis protocols.
a. Version Control and Parameter Tracking
Document all software versions, parameter settings, and database versions used in your analysis. Tools like Conda and Docker enable creation of reproducible computational environments that can be shared and deployed across different systems.
Workflow management systems like Nextflow and Snakemake automatically track parameter settings and software versions while enabling parallel processing across multiple compute nodes. These systems reduce analysis time while ensuring complete reproducibility.
b. Collaborative Analysis Platforms
Modern analysis platforms facilitate collaboration by providing shared workspaces, version control, and standardized analysis protocols. Galaxy, Bioconductor, and specialized platforms enable seamless collaboration between computational and experimental researchers.
By adopting these best practices, you can overcome common bottlenecks, streamline your workflow, and achieve faster, more reliable results.
Conduct Fast RNA Sequencing with Biostate AI
RNA sequencing is an essential tool for understanding gene expression, but the process comes with its challenges. High costs, long wait times, and complex data handling can slow down research progress. In addition, working with low-quality samples and navigating complicated bioinformatics workflows can add unnecessary hurdles.
Biostate AI takes care of all these issues. Our platform is designed to save you time, reduce costs, and make your work easier, so you can focus on what really matters: your research.
Why Choose Biostate AI?
- Unbeatable Pricing: Get high-quality sequencing results starting at $80 per sample, making top-tier RNA sequencing affordable.
- Rapid Turnaround: Receive your results in just 1–3 weeks, enabling faster decision-making and more efficient research.
- Complete Transcriptome Insights: Comprehensive RNA-Seq covering both mRNA and non-coding RNA for deeper biological insights.
- AI-Driven Analysis: Harness the power of OmicsWeb AI for intuitive, actionable insights from your RNA data.
- Minimal Sample Requirement: Process as little as 10µL blood, 10ng RNA, or 1 FFPE slide, offering unparalleled flexibility for varied research needs.
- Low RIN Compatibility: Biostate AI can work with RNA samples having RIN as low as 2 (vs. the typical requirement of ≥5), enabling analysis of valuable, but degraded, clinical samples.
- OmicsWeb: A comprehensive, AI-ready OmicsWeb that automates workflows and prepares your data for further analysis, with no coding expertise required.
Biostate AI offers a complete solution for RNA sequencing, handling every step from sample collection to final insights. This process allows researchers to focus on their studies of cells or organisms without having to manage the complexities and time-consuming aspects of experimental work and lab procedures.
Final Words
Fast, high-quality RNA sequencing analysis demands careful planning, robust workflows, and expert interpretation. However, the complexity of RNA sequencing analysis has led to the development of comprehensive service platforms that handle every aspect of the workflow. These integrated fast RNA sequencing analysis tips eliminate traditional bottlenecks while maintaining scientific rigor.
Biostate AI represents this evolution by providing complete RNA sequencing solutions from sample collection to final insights. Our platform processes diverse sample types, including blood, tissue, culture, and purified RNA, accommodating various research needs with flexibility and precision.
The combination of competitive pricing (starting at $80 per sample), rapid turnaround (1-3 weeks), and comprehensive analysis makes our high-quality RNA sequencing accessible to more researchers.
Ready to see how Biostate AI can advance your research? Request a personalized demo today!
Frequently Asked Questions
- What is the minimum RNA quality required for successful sequencing?
Traditional RNA sequencing protocols require RNA Integrity Numbers (RIN) of 8 or higher for optimal results. However, modern library preparation techniques have expanded compatibility significantly. Biostate AI’s advanced protocols can process samples with RIN values as low as 2, compared to the typical industry requirement of RIN ≥ 5. This breakthrough enables analysis of valuable clinical samples, including FFPE tissues and degraded specimens that would otherwise be unusable.
- How much sequencing depth do I need for my experiment?
Sequencing depth requirements vary significantly based on your research objectives:
- Standard gene expression profiling: 20-30 million mapped reads per sample
- Small RNA sequencing: 5-10 million reads per sample
- Comprehensive isoform analysis: 100+ million reads per sample
- Single-cell RNA sequencing: 50,000-100,000 reads per cell
- Allele-specific expression: 50-100 million reads per sample
The Broad Institute’s analysis of over 10,000 samples demonstrated that 20 million reads capture 95% of expressed genes in human tissues, making this depth optimal for most applications.
- What sample types can be used for RNA sequencing?
RNA sequencing accommodates diverse sample types, each with specific considerations:
- Fresh-frozen tissues: Provide highest RNA quality but require immediate processing
- Blood samples: Convenient but require globin depletion due to high globin mRNA content (up to 80%)
- Cell cultures: Offer controlled conditions and high RNA quality
- FFPE tissues: Enable retrospective studies but introduce RNA fragmentation
- Purified RNA: Allows maximum flexibility but requires careful quality assessment
Biostate AI’s platform demonstrates exceptional versatility by processing samples as small as 10µL blood, 10ng RNA, or single FFPE slides while maintaining analytical accuracy.