
The global DNA next-generation sequencing (NGS) market has grown rapidly due to technology demand. According to Statista, the market is expected to rise from $9.2 billion in 2023 to $66 billion in 2033. This rapid expansion reflects the increasing adoption of NGS technology in medical applications and shows its promising future.
NGS sequencing has contributed greatly to the decoding of genetic information with its faster, more accurate, and cost-effective abilities. But how does an NGS sequencing workflow actually work? Understanding the step-by-step process is crucial for researchers, bioinformaticians, and clinicians who rely on this technology for everything from disease diagnostics to evolutionary studies.
The next generation sequencing workflow involves multiple critical steps, including sample preparation, library construction, sequencing, and data analysis. In this guide, you’ll walk through the entire NGS sequencing process to help you understand how raw DNA transforms into valuable insights. Let’s uncover it below!
What is Next Generation Sequencing (NGS)?
Next generation sequencing (NGS) is a great leap in genomic analysis as it replaced the traditional long-dominant automated Sanger sequencing method. While Sanger sequencing was instrumental in landmark achievements like the first complete human genome, it has certain limitations that are overcome by NGS sequencing.
NGS overcomes the limitations by allowing the researchers to sequence entire genomes, analyze transcriptomes, and study genetic variations without prior knowledge of specific genes. NGS offers a range of sequencing methods that rely on a combination of template preparation, sequencing and imaging, genome alignment, and assembly techniques.
Next generation DNA sequencing has several advantages, but one of the major ones is the ability to generate vast amounts of sequencing data at a much lower cost. Various NGS platforms are available, each suited for different applications. Companies like Roche/454, Illumina, and Life/APG have developed commercial sequencing technologies, with newer entrants like Pacific Biosciences aiming to bring innovative approaches to the market.
Below, you will find the first step in the next-generation DNA sequencing workflow. Let’s explore it.
Step 1: Sample Collection and Preparation (Pre-Analytical Phase)
The first step in a next-generation sequencing workflow is part of a pre-analytical phase. This stage involves proper sample collection, documentation, and handling to ensure sample quality. It is important to remember that the quality, format, and quantity of the sample must be carefully evaluated and documented. Replace the samples that do not meet the minimum quality requirements and replace them with a new one that meets the quality standards.
Sample Characterization for Quality Control (QC)
To ensure sample quality, there are five critical sample characterization questions:
- Source Identification: Determine the original sample source (e.g., blood, tissue, cell culture).
- Isolation Method: Identify the DNA/RNA extraction method used.
- Quantification Method: Specify whether Qubit or NanoDrop was used for concentration assessment.
- Quality Assessment: Confirm whether gel electrophoresis was used for integrity evaluation.
- Clean-Up Procedures: Check if the sample underwent a purification step before shearing to remove contaminants.
Proper sample preparation and documentation of these parameters are crucial for maintaining high-quality sequencing outcomes. Now, below, you’ll explore the nucleic acid stage, which involves tips to ensure the quality of the sample.
Step 2: Nucleic Acid Extraction
At this stage, it is important to ensure that the high-quality DNA/RNA extraction begins. To start this isolation and purification process is important. One of the critical things is temperature control, making sure that these incubation steps are conducted below 60°C, as higher temperatures can degrade nucleic acids. However, it is important to remember that extraction methods can vary based on sample type for example, in certain microbial DNA extractions, the protocol may vary slightly, especially for high-temperature-resistant organisms.
Additionally, to prevent degradation, DNA and RNA samples should be stored on ice whenever possible. Remember to avoid repeated freeze-thaw cycles because they can cause fragmentation and loss of sample integrity. So, fragmenting the sample into smaller volumes can help prevent degradation due to freeze-thawing. Inhibit nuclease activity using appropriate buffer conditions, ensuring that extracted nucleic acids remain stable throughout the process.
Let’s explore how to prepare a library and why this library preparation is important.
Step 3: Library Preparation
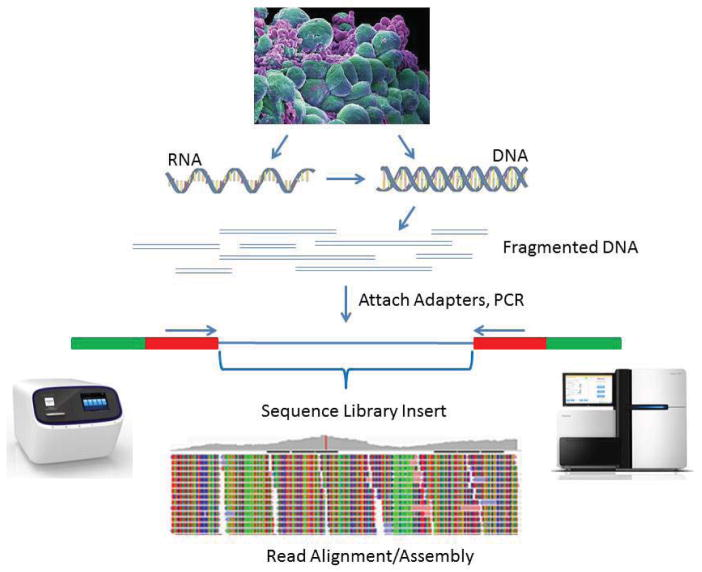
Source: NIH
The construction of a sequencing-ready library from nucleic acid targets (RNA or DNA) is fundamental to the next generation DNA sequencing (NGS) workflow. This process transforms extracted nucleic acids into a format compatible with sequencing platforms such as Illumina, Ion Torrent, Life Technologies, Roche, and Pacific Biosciences.
Key Steps in NGS Library Preparation
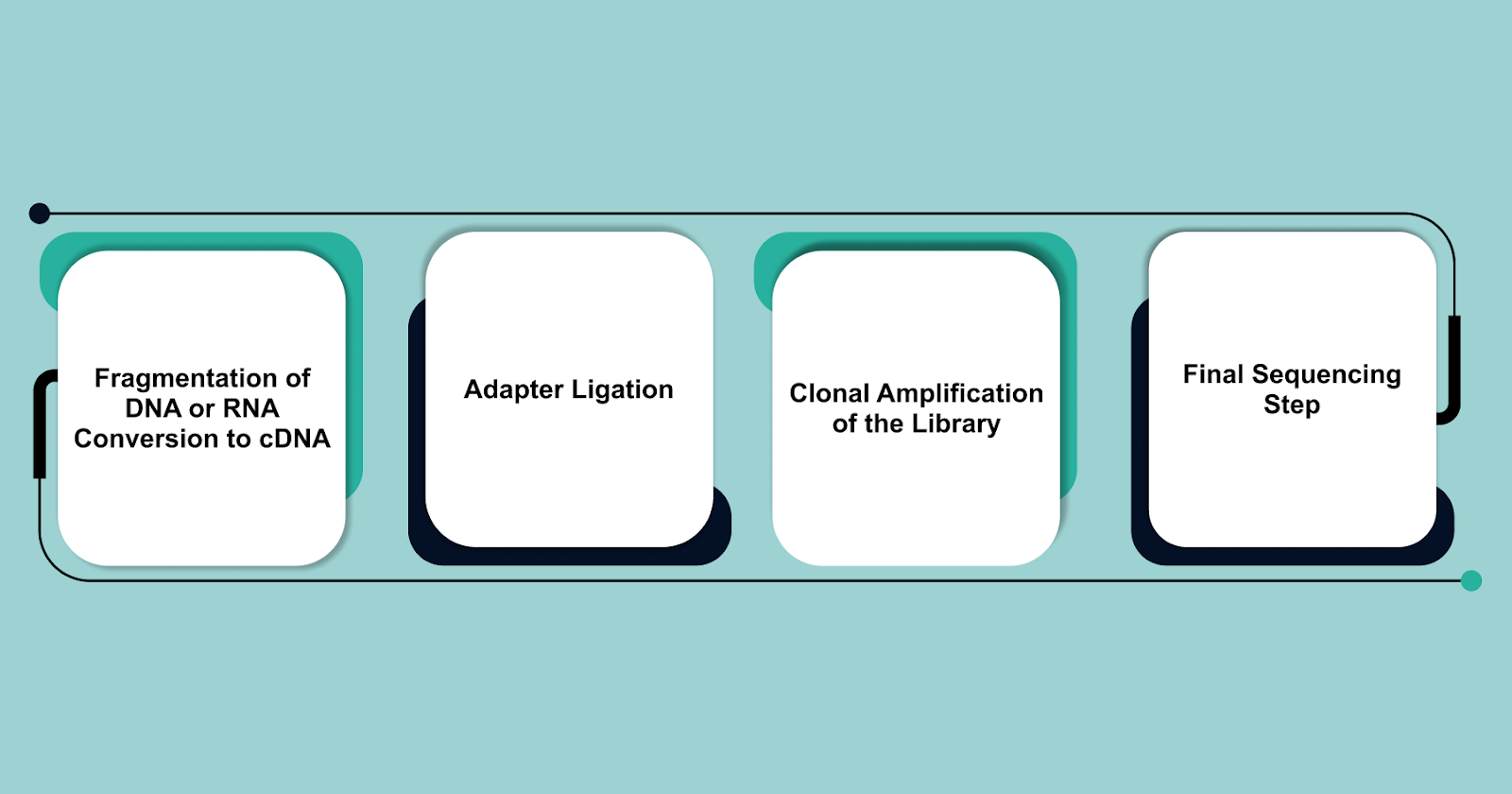
- Fragmentation of DNA or RNA Conversion to cDNA: DNA is fragmented to create appropriate parts for the sequencing. RNA is reverse-transcribed into complementary DNA (cDNA) before fragmentation.
- Adapter Ligation: DNA or cDNA fragments are tied to sequencing adapters that contain platform-specific sequences designed to interact with the sequencing system. In Illumina sequencing, adapters bind to the flow cell surface, whereas in Ion Torrent sequencing, they attach to beads.
- Clonal Amplification of the Library: Cluster generation (Illumina sequencing) or microemulsion PCR (Ion Torrent sequencing) amplifies DNA to ensure detectable sequencing signals. To create clusters on a flow cell, Illumina uses bridge amplification. While Ion Torrent and PacBio use different amplification strategies (like emulsion PCR and SMRT technology, respectively).
- Final Sequencing Step: The prepared next generation DNA sequencing library undergoes sequencing using platform-specific chemistry. One advantage of Illumina sequencing is the ability to perform paired-end sequencing, which enables sequencing from both ends of the library insert for greater accuracy and coverage.
By properly preparing and validating NGS sequencing libraries, researchers ensure accurate, high-quality sequencing that minimizes errors and bias. As you explore the fundamental step of NGS sequencing below, you’ll explore the third-generation sequencing contribution and why it is needed.
Step 4: Third-Generation Sequencing Systems
The fourth step of the next generation sequencing workflow is third-generation sequencing platforms (TGS). This step is included because, though TGS is not always included in NGS workflows, it is a direct evolution of sequencing technologies. It is increasingly used for applications requiring long reads.
The TGS platform Pacific Biosciences RS II has stricter sample quality requirements due to the lack of DNA amplification, which should be known if next generation DNA sequencing is performed.
The following conditions must be met:
- DNA must be in a double-stranded format to ensure optimal sequencing performance.
- Extreme pH levels (below pH 6 or above pH 9) must be avoided.
- The sample should be free from chelating agents, detergents, divalent metal cations, denaturants, and RNA contaminants.
Spectrophotometric analysis is required to assess nucleic acid purity. Additionally, capillary gel electrophoresis should be performed before library preparation to confirm DNA quality. Now, below, you’ll explore how to ensure the quality of the RNA samples.
Step 5: Quality Assessment for FFPE RNA Samples
This quality assessment for FFPE RNA samples is important to include in Next-Generation Sequencing (NGS) because the quality of RNA directly impacts the accuracy and reliability of sequencing results. In formalin-fixed, paraffin-embedded (FFPE) RNA samples, traditionally, the RNA Integrity Number (RIN) has been used to evaluate sample quality based on the 28S to 18S ribosomal RNA peak ratio.
However, an alternative and more reliable measure is DV200, which assesses the percentage of RNA fragments longer than 200 nucleotides. A high DV200 percentage indicates better RNA quality, while a low percentage suggests significant degradation. This metric is considered more reliable than RIN in evaluating degraded RNA samples.
Two main devices can be used for assessing sample quality, such as capillary gel electrophoresis and fluorometric quantification. Most sequencing protocols recommend at least one of these methods for quality control (QC). However, for enhanced precision, both qPCR (quantitative PCR) and ddPCR (digital droplet PCR) can be used for final library quantification.
Below, you will explore the next major step after quality assurance, which is the pipeline for NGS data analysis. Let’s explore it.
Step 6: Bioinformatics Pipeline for NGS Data Analysis
Once sequencing is complete, raw data is stored in standard file formats for processing. The most widely used format is FASTQ, which contains sequence reads along with Phred quality scores.
Other standard file formats include:
- Variant Call Format (VCF): Stores genetic variation data, such as SNPs and Indels.
- Sequence Alignment/Map (SAM) & Binary Alignment/Map (BAM): Used for read alignment against reference genomes.
For data validation, tools like FASTQC, NGS QC Toolkit, and ChromaPipe can evaluate sequence quality, while the Genome Analysis Toolkit (GATK) is widely used for variant calling and genome annotation.
You have landed on the very last step of NGS sequencing, which is data submission and storage guidelines. Let’s explore it below.
Step 7: Data Submission and Storage
Once QC, analysis, and annotation are complete, sequencing data must be submitted to public databases or customers. To meet minimum data submission requirements, sequencing coverage should be at least 90%, and gaps should be minimized through gap resolution techniques.
The Genomic Standards Consortium (GSC) introduced the Minimum Information about a Genome Sequence (MIGS) specification, which ensures that sequencing data includes biological context details, such as taxonomy and trophic level.
For high-quality sequencing, the Bermuda Standards specify that data should be 99.99% accurate (one error per 10,000 base pairs), with fully assembled sequences containing no missing gaps. This accuracy applies primarily to finished assemblies, while raw sequencing data typically has more flexibility in accuracy thresholds (though still stringent).
You have covered all the major steps needed to understand the NGS sequencing workflow. Below is a summary of the steps for successful sequencing and data analysis.
Conclusion
NGS workflows involve strict sample preparation, quality control, sequencing, and data analysis protocols. By following this procedure for DNA/RNA extraction, sample quality assessment, bioinformatics processing, and data submission, the researcher can accurately achieve reproducible sequencing results. As NGS continues to evolve, you can expect improved accuracy, faster turnaround times, and reduced costs. Innovations like single-cell sequencing, real-time data analysis, and better integration with AI will enhance its utility.
The continued growth of NGS technology will open doors to new applications. As NGS sequencing continues to evolve, decoding genetic information and human understanding will reach new heights. Many platforms like Biostate.ai are dedicated to increasing the understanding of genes and RNA at an affordable cost. Biostate.ai offers full RNA sequencing insights at a cost-effective rate that is both accurate and high-quality. Book Your Consultation Today!
Disclaimer: This article provides general information about next-generation sequencing workflow. It is not intended as medical advice. For any medical concerns, always consult with a licensed healthcare professional.
FAQ
- What is the first step in the NGS workflow, and why is it important?
Answer: The first step in the NGS workflow is sample collection and preparation. This step is crucial, as proper sample documentation and management are important to maintaining its quality. Researchers need to make sure of the right sample type (e.g., blood, tissue, or cell culture) and must assess the quality using methods like gel electrophoresis and concentration tests. Poor sample preparation can lead to degraded or inaccurate data, compromising the entire sequencing process.
2. What role does library preparation play in NGS, and what key steps are involved?
Answer: Library preparation is a critical and fundamental part of NGS sequencing because it converts the extracted nucleic acids (DNA or RNA) into a format that sequencing platforms can read. The key steps involved are:
- Fragmentation: DNA or RNA is broken into smaller pieces suitable for sequencing.
- Adapter Ligation: Sequencing adapters are attached to the fragments to enable them to bind to the sequencing platform.
- Amplification: The library undergoes amplification to generate sufficient amounts of DNA for sequencing.
3. What is the significance of third-generation sequencing (TGS) in the NGS workflow?
Answer: Third-generation sequencing (TGS) is added to the procedure because it is used for long reads and can help resolve the genomic regions. There are platforms like Pacific Biosciences (PacBio) and Oxford Nanopore that offer advantages over traditional NGS by providing more comprehensive coverage of the genome, especially for studying structural variants and repetitive regions. TGS does not require DNA amplification, making it ideal for sequencing large genomes or challenging samples such as those from FFPE (formalin-fixed paraffin-embedded) tissues.
4. Why is sample quality assessment critical, particularly for RNA samples?
Answer: Quality assessment of RNA samples is crucial because RNA degradation can significantly affect the accuracy of sequencing results. Samples from FFPE tissues are prone to RNA degradation. The RNA Integrity Number (RIN) was traditionally used to measure the quality of RNA. Still, a more reliable method is the DV200, which assesses the percentage of RNA fragments longer than 200 nucleotides. Ensuring high RNA quality allows for more accurate data interpretation and reduces the risk of errors in downstream analysis.
5. What happens after sequencing is completed in the NGS workflow?
Answer: After sequencing, the raw data undergoes analysis using bioinformatics tools. The data is typically stored in FASTQ format, and various stages of data analysis occur:
- Base calling: Identifying individual nucleotides.
- Filtering: Removing low-quality or erroneous sequences.
- Alignment: Mapping the sequences to a reference genome.
- Variant calling: Identifying genetic variations like SNPs or indels. The results are then interpreted to predict gene functions, pathways, and other biological insights.
- Finally, data must be submitted to public databases or customers according to submission standards like those set by the Bermuda and Genomic Standards Consortium.