
RNA-Seq can reveal several unique insights into gene expression that other methods miss. RNA sequencing (RNA-Seq) has become an important tool for scientists, offering a comprehensive view of the transcriptome and allowing deeper exploration of gene activity than ever before.
Unlike older methods such as Sanger sequencing or microarrays, RNA-Seq offers far greater depth and precision in studying gene expression, alternative splicing, and even allele-specific expression. Its applications extend beyond traditional mRNA quantification to include the analysis of noncoding RNA, such as microRNA and long noncoding RNA.
This article provides a comprehensive view of the importance of RNA-Seq in modern research, its evolving workflow, and how it enables detailed transcriptome analysis across various biological contexts. By embracing RNA-Seq, researchers can uncover novel genetic insights, which are shaping the future of molecular biology and personalized medicine.
Preparation for RNA-Seq Data Analysis
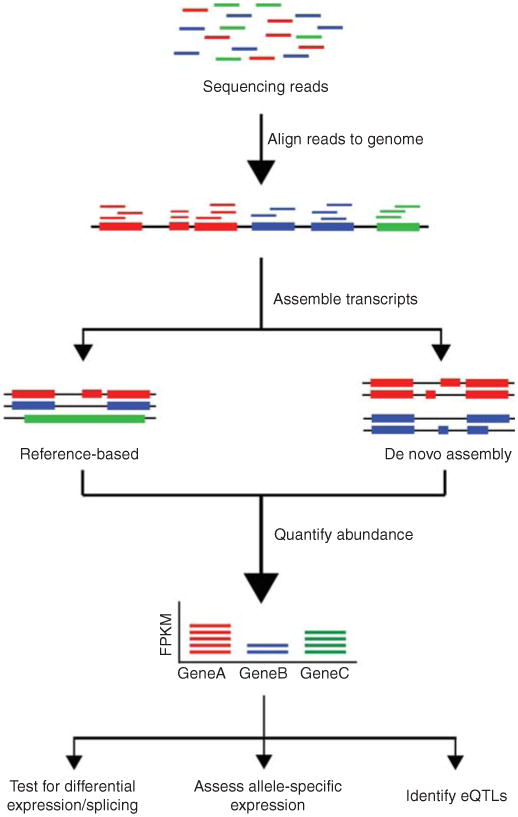
Sources: NIH Overview of RNA-Seq data analysis
The procedure for RNA sequences begins with the preparation of raw data in paired-end FASTQ format from the sequencing facility. These files (e.g., read_1.fq.gz and read_2.fq.gz) contain the sequences generated during sequencing. Before proceeding with further analysis, it’s crucial to verify that the data is of high quality. This is done through initial quality control (QC) to assess if the sequencing data contains any issues that could impact subsequent steps.
Below is the quality control of RNA-seq data, which ensures that the results are reliable and suitable for downstream analysis.
Quality Control of RNA-Seq Data
Quality control is a very important step in evaluating the overall quality and integrity of the raw-seq data. Tools like FastQC are used to assess the quality of the sample, GC content distribution, and overrepresented sequences. After running FastQC, you’ll obtain a comprehensive report highlighting potential issues in your raw sequencing data. Key metrics include per-base sequence quality, per-sequence GC content, and adapter contamination.
These metrics help you decide whether further preprocessing steps, such as trimming or filtering, are necessary before downstream analysis. It also provides reports that include base-level sequence quality, GC content at each position in the reads, and checks for contamination by rRNA or adapters. These reports can indicate whether the data needs any cleaning or is ready for the next steps. Common issues that might arise include low-quality bases at the ends of reads or the presence of rRNA contamination, which would need to be filtered out.
Below are the read trimming and alignment steps, which are necessary steps in RNA-seq analysis to ensure the data is of high quality and can be accurately mapped to a reference genome or transcriptome for further study.
Read Trimming and Alignment
After initial QC, the next step is to process the data by trimming or removing the low-quality bases from the 3’ end of the reads and removing any adapter sequences that may be present. This is important to improve the quality of the data for accurate alignment. Tools like Trimmomatic can be used for this purpose, with parameters set to trim reads based on a specific quality threshold (e.g., Phred score of 20).
Proper trimming hugely enhances the accuracy of read alignment by removing noisy or contaminant sequences that can lead to mismapped reads or false positives. It also reduces the computational burden and improves the overall signal-to-noise ratio in the dataset. During this step, it’s essential to generate post-trimming QC reports using FastQC or MultiQC again to confirm the effectiveness of trimming and verify improvements in quality metrics like read length distribution and base quality scores.
Ensuring the removal of adapter sequences and low-quality tails is especially critical in RNA-seq, where misaligned reads can distort expression profiles and compromise the biological interpretations. After trimming, the clean reads are aligned to a reference genome or transcriptome using alignment tools like STAR or Bowtie. The alignment ensures that each read is mapped to the correct genomic location, which is crucial for downstream analysis, such as gene expression quantification.
After this step, below is the counting and normalization of gene expression, which is an important step as it allows you to quantify and compare the levels of gene expression across different samples or conditions.
Counting and Normalizing Gene Expression
Once the reads are aligned, the next step is to quantify the gene expression levels by counting the number of reads that map to each gene or transcript. This is typically done using tools like HTSeq or featureCounts, which create a count table showing how many reads correspond to each gene. Before proceeding to normalization, it’s important to carefully inspect the count data for potential anomalies such as genes with zero counts across all samples or unusually high counts in a single sample, which may indicate mapping errors or batch effects. Filtering out lowly expressed genes at this stage can improve statistical power and reduce noise in the data.
Tools like DESeq2 and edgeR often include built-in functions for such filtering, helping to streamline the workflow and prepare the dataset for meaningful differential expression analysis. These raw counts are then normalized to adjust for technical biases, such as variations in sequencing depth across samples. Then, using normalization methods, like the Variance Stabilizing Transformation (VST) in DESeq2 or edgeR, ensure that gene expression comparisons between different conditions are accurate and reflect biological differences.
After this essential step, differential expression analysis is implemented to understand how gene activity changes under different conditions.
Differential Expression Analysis
Differential expression (DE) analysis is the next step, where researchers aim to identify genes or transcripts that show significant changes in expression between experimental conditions. Using tools like DESeq2 or edgeR, researchers can perform statistical analysis to determine which genes are upregulated or downregulated under specific conditions. To perform DE analysis effectively, it’s essential to provide well-designed metadata describing sample conditions, replicates, and batch information.
This allows tools like DESeq2 and edgeR to accurately model the experimental design using generalized linear models or negative binomial distributions. The output typically includes fold-change values, p-values, and adjusted p-values (FDR) to assess statistical significance. Visualizations such as MA plots, volcano plots, and heatmaps are often generated to help interpret the results and highlight key genes of interest. These tools account for the inherent variability in the data and identify genes that are consistently differentially expressed, providing valuable insights into the biological processes under study.
Below, you will find the last step of RNA-seq data analysis, visualization, which allows researchers to interpret complex gene expression patterns more easily.
Visualization of RNA-Seq Data
Visualization of RNA-Seq data is a critical step in interpreting the results. Tools like principal component analysis (PCA) and heatmaps help researchers visualize patterns in gene expression across samples. PCA allows for the exploration of how different conditions cluster or separate, providing a quick overview of potential biological patterns or outliers. Addition to PCA and heatmaps, other visualization techniques such as hierarchical clustering, boxplots, and gene-specific expression plots (e.g., using tools like ggplot2 or pheatmap in R) can offer deeper insights into expression trends.
For example, volcano plots are commonly used to display the relationship between fold change and statistical significance, making it easier to identify the most impactful differentially expressed genes. Effective visualization not only supports data interpretation but also ensures reproducibility and clarity in scientific communication, especially when preparing figures for publications or presentations. Heatmaps can show gene expression patterns across multiple samples, highlighting genes that behave similarly. These visualizations help researchers understand the biological relevance of their findings and communicate results effectively.
Above, you have covered all the steps that are important in the RNA-seq data analysis process. Below, you will find a quick overview of what you have learned.
Conclusion
RNA-Seq data analysis is a powerful approach to understanding gene expression at a deep level, offering insights into gene activity, alternative splicing, and noncoding RNAs. Researchers can extract meaningful biological information by following a structured workflow—starting with quality control, moving through read trimming, alignment, expression quantification, differential expression analysis, and ending with visualization.
This analysis enables the discovery of novel genetic insights, contributing significantly to fields like molecular biology and personalized medicine. By mastering RNA-Seq, scientists can enhance their research and uncover previously hidden patterns in gene expression. With continuous advancements in computational tools and sequencing technologies, RNA-Seq has become more accessible and scalable. Whether analyzing a few samples or hundreds, a well-executed RNA-Seq workflow ensures reliable results that can drive hypothesis generation, biomarker discovery, and therapeutic development.
If you are a researcher looking to unlock deeper insights from your RNA-Seq data, then choose Biostate.ai. It offers comprehensive insights and delivers high-quality RNA sequencing solutions at an affordable price. Supporting global researchers, its services cater to a wide range of research needs, from studying cells and tissues to exploring diseases. Get Your Quote Now!
Disclaimer: The information provided in this article is for educational and informational purposes only. While every effort has been made to ensure the accuracy and reliability of the content, it is recommended to consult with experts or professionals in the field of RNA sequencing for specific research or experimental advice.
FAQs
- What is the RNA-Seq data analysis method?
RNA-Seq data analysis is a computational method that processes raw RNA sequencing data to study gene expression, detect alternative splicing events, and identify and quantify non-coding RNAs through steps like quality control, alignment, read counting, and differential expression analysis.
- How to analyze RNA-Seq raw data?
To analyze RNA-Seq data, start with quality control using tools like FastQC, then trim low-quality bases and adapters with Trimmomatic. Align clean reads to a reference genome using STAR or HISAT2. Count mapped reads with HTSeq or featureCounts, followed by normalization and statistical analysis for expression comparison across samples.
- What is the workflow of RNA-Seq data analysis?
RNA-Seq analysis begins with quality control and trimming, followed by alignment using tools like STAR or HISAT2. Alternatively, transcript-level quantification can be performed using alignment-free tools like Salmon or Kallisto. Expression is quantified, differential expression analyzed, and results visualized using PCA, heatmaps, or other methods.
- What is the primary analysis of RNA-Seq?
Primary analysis of RNA-Seq involves quality control, trimming low-quality reads, and aligning sequences to a reference genome or transcriptome. It ensures clean, accurate data for downstream tasks like gene expression quantification and differential analysis, forming the foundation for reliable biological interpretation.